Un fármaco contra el cáncer existente alivia un síndrome genético poco común

Un estudio reciente muestra que ruxolitinib, un fármaco dirigido al interferón gamma (IFN-gamma), alivia eficazmente los síntomas del raro síndrome poliendocrino autoinmune tipo 1 (APS-1) tanto en ratones como en humanos, allanando el camino hacia nuevas tratamientos para esta y otras enfermedades relacionadas. Modelo 3D del fármaco ruxolitinib. Crédito: NIAID
Investigadores del NIH han descubierto nuevas vías que podrían conducir a tratamientos para el síndrome poliendocrino autoinmune tipo 1.
Un medicamento diseñado originalmente para algunos trastornos autoinmunes y el cáncer ha resultado eficaz en el tratamiento de los síntomas de una rara enfermedad genética conocida como síndrome poliendocrino autoinmune tipo 1 (APS-1). Este hallazgo surgió después de que los investigadores descubrieran que el síndrome se correlaciona con niveles elevados de interferón gamma (IFN-gamma), una proteína que desempeña un papel en las respuestas inmunitarias.
Este descubrimiento arroja luz sobre el papel del IFN-gamma en las enfermedades autoinmunes. La investigación, dirigida por el Instituto Nacional de Alergias y Enfermedades Infecciosas, parte del Institutos Nacionales de Saludfue publicado en Revista de medicina de Nueva Inglaterrami.
En un estudio de tres etapas, realizado en ratones y personas, los investigadores examinaron cómo APS-1 causa enfermedades autoinmunes. El síndrome se caracteriza por disfunción orgánica múltiple, suele comenzar en la infancia y es mortal en más del 30% de los casos. Este síndrome hereditario es causado por una deficiencia en un gen que impide que las células T del sistema inmunológico ataquen las células del cuerpo, lo que lleva a la autoinmunidad; infecciones crónicas por hongos en la piel, uñas y membranas mucosas; y producción insuficiente de hormonas por parte de órganos endocrinos, como las glándulas suprarrenales. Los síntomas incluyen irritación del estómago, inflamación del hígado, irritación de los pulmones, caída del cabello, decoloración de la piel, daño a los tejidos e insuficiencia orgánica.
Resultados de investigaciones y pruebas de drogas
En la primera etapa de este estudio, investigadores dirigidos por científicos del Laboratorio de Inmunología Clínica y Microbiología del NIAID examinaron la historia natural de APS-1 en 110 adultos y niños. Se analizaron sangre y tejidos para comparar la expresión de genes y proteínas en personas con y sin APS-1. Encontraron respuestas elevadas de IFN-gamma en la sangre y los tejidos de personas con APS-1, lo que indica que el IFN-gamma puede desempeñar un papel importante en la enfermedad y proporcionar una vía hacia un objetivo para el tratamiento.
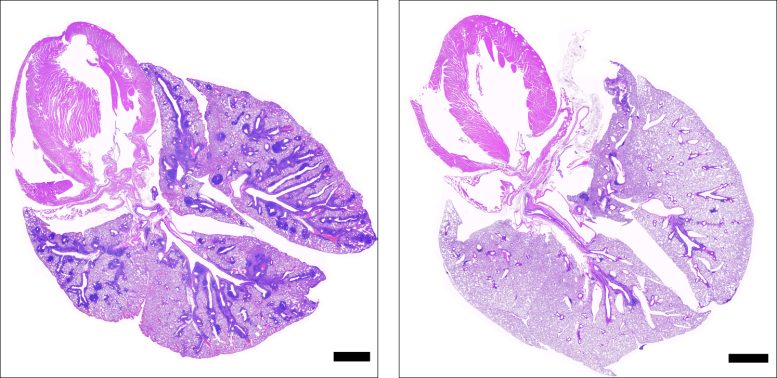
Cortes transversales de pulmones de ratones deficientes en el gen que causa APS-1, que muestran tejido dañado en ratones que no recibieron ruxolitinib (izquierda) y tejido sano en ratones que recibieron ruxolitinib (derecha). Las barras negras representan 1 mm. Crédito: NIAID
En la segunda etapa del estudio, los científicos examinaron ratones con la misma deficiencia genética que causa APS-1 en las personas y descubrieron que los animales también experimentaban daño en el tejido autoinmune y niveles elevados de IFN-gamma. Los ratones también deficientes en el gen de IFN-gamma no tenían daño en el tejido autoinmune, lo que mostró un vínculo directo entre los síntomas de IFN-gamma y APS-1.
Con este conocimiento, los investigadores buscaron un fármaco que pudiera usarse para reducir la actividad del IFN-gamma en personas. Eligieron ruxolitinib, un inhibidor de la Janus quinasa, porque actúa cerrando la vía impulsada por el IFN-gamma. Cuando se administró ruxolitinib a ratones con la deficiencia genética que causa APS-1, las respuestas de IFN-gamma se normalizaron y se impidió que las células T se infiltraran en los tejidos y dañaran los órganos. Estos resultados mostraron que ruxolitinib puede aliviar los efectos de la deficiencia genética, lo que sugiere que puede ser eficaz para el tratamiento del APS-1 en personas.
Los investigadores administraron ruxolitinib, suministrado por el Centro Clínico de los NIH, a cinco personas (dos adultos y tres niños) con APS-1 en la tercera etapa del estudio. Las dosis y los regímenes se adaptaron a cada individuo y los tratamientos continuaron durante más de un año. El fármaco fue seguro y bien tolerado, y se observó una mejoría de los síntomas en todos los participantes del estudio. Los análisis de sangre y tejidos mostraron una reducción en la producción de IFN-gamma por las células T, así como niveles normalizados de IFN-gamma en la sangre. Se redujeron muchos síntomas relacionados con APS-1, incluida la caída del cabello, las infecciones orales por hongos, la irritación estomacal e intestinal, la urticaria y la inflamación de la tiroides.
Los resultados mostraron que la normalización de los niveles de IFN-gamma utilizando ruxolitinib puede reducir los efectos nocivos de APS-1 en las personas. Los científicos señalan que se necesita un estudio con un grupo más grande y diverso de pacientes para determinar si ruxolitinib y medicamentos similares son tratamientos adecuados para personas con APS-1. Escriben que comprender el papel del IFN-gamma en la autoinmunidad puede conducir al desarrollo de tratamientos para enfermedades relacionadas. Esta investigación destaca la importancia de encontrar causas y tratamientos para enfermedades raras.
Referencia: «El papel del interferón-γ en el síndrome poliendocrino autoinmune tipo 1» por Vasileios Oikonomou, Grace Smith, Gregory M. Constantine, Monica M. Schmitt, Elise MN Ferré, Julie C. Alejo, Deanna Riley, Dhaneshwar Kumar, Lucas Dos Santos Dias, Joseph Pechacek, Yannis Hadjiyannis, Taura Webb, Bryce A. Seifert, Rajarshi Ghosh, Magdalena Walkiewicz, Daniel Martin, Marine Besnard, Brendan D. Snarr, Shiva Deljookorani, Chyi-chia R. Lee, Tom DiMaggio, Princess Barber, Lindsey B. Rosen, Aristine Cheng, Andre Rastegar, Adriana A. de Jesus, Jennifer Stoddard, Hye Sun Kuehn, Timothy J. Break, Heidi H. Kong, Leslie Castelo-Soccio, Ben Colton, Blake M. Warner, David E. Kleiner, Martha M. Quezado, Jeremy L. Davis, Kevin P. Fennelly, Kenneth N. Olivier, Sergio D. Rosenzweig, Anthony F. Suffredini, Mark S. Anderson, Marc Swidergall, Carole Guillonneau, Luigi D. Notarangelo, Raphaela Goldbach -Mansky, Olaf Neth, María Teresa Monserrat-García, Justo Valverde-Fernández, José Manuel Lucena, Ana Lucía Gómez-Gila, Ángela García Rojas, Mikko RJ Seppänen, Jouko Lohi, Matti Hero, Saila Laakso, Paula Klemetti, Vanja Lundberg, Olov Ekwall, Peter Olbrich, Karen K. Winer, Behdad Afzali, Niki M. Moutsopoulos, Steven M. Holland, Theo Heller, Stefania Pittaluga y Michail S. Lionakis, 29 de mayo de 2024, Revista de medicina de Nueva Inglaterra.
DOI: 10.1056/NEJMoa2312665
La investigación fue financiada por el Instituto Nacional de Alergias y Enfermedades Infecciosas.
Related Posts

7 ácidos fuertes: cómo darle vida a la química con humor y personalidad7 ácidos fuertes – High School/Honores/AP® Chemistry Resources
Los recursos contra el acoso y el acoso sexual aumentan en las escuelas de EE. UU., pero persisten las brechas
